

ЁЁЁЁЁЁвНСЦЦїаЕзЂВсїХЙиНкМйЬхгІдкТњзузЂВсЗЈЙцвЊЧѓЕФЧАЬсЯТЃЌПЩАДееЁЖвНСЦЦїаЕСйДВЦРМлММЪѕжИЕМддђЁЗНјааЭЌЦЗжжВњЦЗЕФСйДВЪ§ОнЖдБШЁЂЗжЮіЁЂЦРМлЃЌВЂАДееЁЖвНСЦЦїаЕСйДВЦРМлММЪѕжИЕМддђЁЗвЊЧѓЕФЯюФПКЭИёЪНГіОпЦРМлБЈИцЁЃ
ЁЁЁЁЖдгкашвЊНјааСйДВЪдбщЕФЃЌгІЕБАДееЁЖвНСЦЦїаЕСйДВЪдбщжЪСПЙмРэЙцЗЖЁЗЕФвЊЧѓЪЕЪЉЁЃЬсНЛЕФСйДВЦРМлзЪСЯгІЕБАќРЈСйДВЪдбщавщЁЂСйДВЪдбщЗНАИКЭСйДВЪдбщБЈИцЁЃПЊеЙСйДВЪдбщбаОПЪБЃЌдкСйДВЪдбщЗНАИжЦЖЈжаНЈвщПМТЧвдЯТвђЫиЃЌАќРЈЕЋВЛЯогкЃК
ЁЁЁЁ1.СйДВЪдбщЕЅдЊ
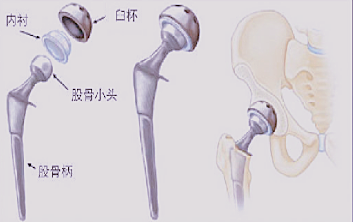
ЁЁЁЁвЛИізЂВсЕЅдЊФкЕФВњЦЗЃЌЩъЧыШЫгІИљОндЄЦкЩъБЈЕФЪЪгУЗЖЮЇВЛЭЌ(ГѕДЮжУЛЛЁЂМйЬхЗаоЁЂжзСіЙиНкжУЛЛ)ЃЌЛЎЗжЮЊВЛЭЌЕФСйДВЪдбщЕЅдЊНјааЪдбщЁЃ
ЁЁЁЁ2.СйДВЪдбщЩшМЦРраЭ
ЁЁЁЁПМТЧВњЦЗЮЊїХЙиНкгРОУадЬцДњжВШыЮяЃЌЪєгкНЯИпЗчЯевНСЦЦїаЕЃЌЮЊСЫБЃжЄЪдбщНсЙћЕФецЪЕПЭЙладКЭПЩБШадЃЌНЈвщВЩгУОпгаСМКУЖдееЕФЧАеАадЕФЫцЛњЖдееСйДВЪдбщЁЃ
ЁЁЁЁШчЙћЩъЧыШЫВЩгУЗЧЫцЛњЦНааЖдеебаОПЃЌдкСЦаЇЦРМлЪБЃЌгаПЩФмгЩгкЛљЯпВЛОљКтЖјЕМжТЮоЗЈПЭЙлЕФЦРМлЪдбщНсЙћЃЌЩъЧыШЫгІЬсЙЉГфЗжЕФРэгЩНтЪЭНсЙћЕФПЭЙладКЭецЪЕадЁЃ
ЁЁЁЁШчгажЄОнБэУїЭЌРрШЋїХЙиНкМйЬхдкЪЕМЪСйДВЪЙгУжаБэЯжСМКУЃЌЧвБОДЮЩъБЈВњЦЗдкЩшМЦМАЩњВњЙЄвеЮДЗЂЩњЪЕжЪБфЛЏЃЌПЩдкСйДВЪдбщЩшМЦжагІгУЕЅзщФПБъжЕЗЈЃЌМДСйДВЪдбщВЛЩшСЂЖдеезщЁЃ
ЁЁЁЁ3.ШыбЁЁЂХХГ§БъзМ
ЁЁЁЁЪмЪдепгІДгашвЊНјааШЋїХЙиНкжУЛЛЪѕжЮСЦЕФвЛАуеяЖЯШЫШКжабЁГіЁЃЩъЧыШЫМАСйДВбаОПЛњЙЙгІИљОнЩъБЈВњЦЗЕФЩшМЦЬиеїМАЦфЪЪгУЗЖЮЇжЦЖЈЦфСйДВЪдбщЕФШыбЁ/ХХГ§/ЭЫГіБъзМЃЌВЛЗћКЯЫљгаШыбЁБъзМЛђепЗћКЯШЮКЮвЛЯюХХГ§БъзМЕФбаОПЖдЯѓгІБЛХХГ§ЁЃОйР§ШчЯТЃК
ЁЁЁЁГѕДЮжУЛЛгУЙЧЫЎФраЭШЋїХЙиНкМйЬхСйДВЪдбщШыбЁБъзМЃК
ЁЁЁЁ(1)ЛМепФъСфдк18—80ЫъЃЌадБ№ВЛЯоЃЌЕЭгк50ЫъЕФЛМепгІгаЪжЪѕЕФНєЦШадЫпЧѓЁЃ
ЁЁЁЁ(2)ЛМепЙЧїРвбГЩЪьЁЃ
ЁЁЁЁ(3)ЛМепгІОпгаШЋїХЙиНкжУЛЛЪжЪѕЪЪгІжЄЁЃР§ШчЃКдЗЂадЭЫБфадїХЙиНкЙЧЙиНкбзЁЂРЯФъЙЩЙЧОБЭЗЯТаЭЛђЭЗОБаЭЙЧелЁЂЙЩЙЧЭЗШБбЊЛЕЫРНјШыЕкIVЦкЁЂїХОЪЗЂг§ВЛСМЫљжТЕФїХЙиНкЙЧЙиНкбзЁЂРрЗчЪЊЙиНкбзЛђЧПжБадМЙжљбзЕШШЋЩэМВВЁїХЙиНкЪмРлЁЂЗЂг§адїХЭбЮЛЛМепїХВПбЯжиЬлЭДМАЛюЖЏеЯАЁЂїХВПДДЩЫКѓЙЧЙиНкбзЁЂГТОЩадїХОЪЙЧелЁЂФбвдСМКУЪжЪѕИДЮЛЕФїХЙиНкФкЙЧелЁЃ
ЁЁЁЁ(4)ЪмЪдВрЛМжЋЮЊГѕДЮНгЪмШЋїХЙиНкжУЛЛЁЃ
ЁЁЁЁ(5)ЪѕЧАЃЌЪмЪдепЛђМрЛЄШЫдИвтВЂЧвФмЙЛЧЉЪ№жЊЧщЭЌвтЪщЁЃ
ЁЁЁЁХХГ§БъзМЃК
ЁЁЁЁ(1)ЛМепЩёОМЁШтЙІФмВЛШЋ(Р§ШчЃКТщБдЁЂМЁШмНтЛђЭтеЙМЁЮоСІ)ЛсЕМжТЪѕКѓїХЙиНкВЛЮШЖЈЛђВНЬЌвьГЃЁЃ
ЁЁЁЁ(2)ЛМепОЋЩёЩЯЮоФмСІЛђепВЛФмРэНтВЮгыбаОПЕФвЊЧѓЁЃ
ЁЁЁЁ(3)аяОЦепЛђЮќЖОепЁЂвЉЮяРФгУепЁЃ
ЁЁЁЁ(4)дЄМЦвРДгадВюЁЃ
ЁЁЁЁ(5)ЗЪХжBMI>35ЁЃ
ЁЁЁЁ(6)вбжЊЛМепЖдвЛжжЛђЖржжжВШыЕФВФСЯгаЙ§УєЪЗЁЃ
ЁЁЁЁ(7)їХЙиНкЛђЩэЬхЦфЫћВПЮЛДцдкЛюЖЏадИаШОВЁдюЁЃ
ЁЁЁЁ(8)їХЙиНкжмЮЇОпгабЯжиЕФЙЧжЪЪшЫЩЁЂДњаЛадЙЧВЁЁЂЗХЩфадЙЧВЁЁЂжзСіЁЃ
ЁЁЁЁ(9)даИОЛђВИШщЦкИОХЎЁЃ
ЁЁЁЁ(10)їХЙиНкЗЂг§ВЛСМCROWEЗжМЖ3ЁЂ4МЖЁЃ
ЁЁЁЁ(11)ЬхжЪащШѕЛђвђШЋЩэЦфЫћМВВЁВЛФмФЭЪмЪжЪѕепЃЌдЄЦкЪйУќВЛзу2ФъЁЃ
ЁЁЁЁ(12)ЪмЪдепКЯВЂЕФЦфЫћМВВЁЯожЦЦфВЮМгбаОПЃЌВЛФмвРДгЫцЗУЛђгАЯьбаОПЕФПЦбЇадЭъећадЁЃ
ЁЁЁЁ(13)ЪмЪдепШыбЁЧАдјВЮМгЙ§ЦфЫћвЉЮяЁЂЩњЮяжЦМСЛђвНСЦЦїаЕСйДВбаОПЖјЮДДяЕНжївЊбаОПжеЕуЪБЯоепЁЃ
ЁЁЁЁЖдгкЗаоаЭїХЙиНкМйЬхЁЂжзСіаЭїХЙиНкМйЬхЕШВњЦЗЃЌгІИљОнВњЦЗЬиЕужЦЖЈШыбЁБъзМКЭХХГ§БъзМЁЃ
ЁЁЁЁ4.ЪмЪдепЭЫГіБъзММАЭЫГіЪмЪдепЕФДІРэ
ЁЁЁЁЭЫГіБъзМ
ЁЁЁЁ(1)ЪмЪдепГЗЛижЊЧщЭЌвтЪщ;
ЁЁЁЁ(2)бЯжиЮЅЗДСйДВЪдбщЗНАИ;
ЁЁЁЁ(3)баОПепШЯЮЊВЛдйЪЪКЯМЬајНјааСйДВЪдбщеп;
ЁЁЁЁ(4)дкСйДВЪдбщЦкМфШбЩяЕФИОХЎ;
ЁЁЁЁ(5)ЪмЪдепЫРЭі;
ЁЁЁЁ(6)ЪмЪдепЪЇЗУ;
ЁЁЁЁ(7)ЩъЧыШЫвЊЧѓжежЙСйДВЪдбщЁЃ
ЁЁЁЁЭЫГіЪмЪдепЕФДІРэ
ЁЁЁЁ(1)ЫљгаЭЫГіЪмЪдепОљгІБЃСєШЋВПдДЪ§ОнКЭдДЮФМўЁЃЕБЪмЪдепЭЫГіКѓЃЌбаОПепгІВЩШЁЖржжаЮЪНШчЕчЛАЁЂгЪМўЕШОЁПЩФмгыЪмЪдепСЊЯЕЃЌбЏЮЪРэгЩ;
ЁЁЁЁ(2)НЋжежЙСйДВЪдбщЕФЪБМфКЭдвђЯъЯИМЧТМдкВЁР§БЈИцБэЩЯ;
ЁЁЁЁ(3)вђВЛСМЪТМўЖјЭбТфепШчОЫцЗУзюжеХаЖЯгыЪдбщЦїаЕДцдквђЙћЙиЯЕЃЌБиаыМЧТМдкCRFБэжаЃЌВЂЭЈжЊЩъЧыШЫЁЃЖдвђВЛСМЪТМўЖјжежЙЪдбщЕФВЁШЫБиаыЫцЗУжСВЛСМЪТМўЕУЕННтОіЛђЮШЖЈЁЃ
ЁЁЁЁ(4)ЁЖвНСЦЦїаЕСйДВЪдбщжЪСПЙмРэЙцЗЖЁЗЙцЖЈЕФЦфЫћЯрЙиЪТвЫЁЃ
ЁЁЁЁ5.СйДВЪдбщГжајЪБМфгыДАПкЦк
ЁЁЁЁСйДВЪдбщЕФГжајЪБМфШЁОігкЫљгаАВШЋадКЭгааЇадЪ§ОнЕФЛёЕУЃЌбаОПВЁР§жСЩйЫцЗУжС12ИідТвдЩЯЁЃНЈвщдкЪжЪѕЧАЁЂЪѕКѓЁЂ6жмЁЂ3ИідТЁЂ6ИідТЁЂ12ИідТЕФїХЙиНкжУЛЛМйЬхЯЕЭГЕФСйДВбаОПЪ§ОнНјааЪеМЏЁЃШчЪѕКѓМДПЬЕФXЩфЯпЦНЦЌЃЌУПДЮЫцЗУНЈвщАќРЈЛМепжїЫпЁЂЬхИёМьВщЁЂXЯпЦНЦЌЁЂЙиНкЙІФмЦРЗжЁЂвдМАжИЕМЛМепЙІФмПЕИДЕШФкШнЁЃ
ЁЁЁЁ6.СйДВЪдбщЦРМлжИБъМАХаЖЈБъзМ
ЁЁЁЁСйДВЪдбщФЩШыВЁР§вЛАугІЮЊЕЅВрїХЙиНкжУЛЛЃЌШчВЁЛМашааЫЋВржУЛЛЃЌгІдкЕЅВржВШыжСЩй3ИідТКѓЃЌОЙ§ЦРМлВЛЛсЖдСэЭтвЛВрВњЩњгАЯьЃЌЗНПЩЪЉааїХЙиНкжУЛЛЁЃЗёдђЫЋВржУЛЛВЁР§гІбЁШЁСЦаЇЯрЖдВюЕФвЛВрНјааЦРМлЁЃ
ЁЁЁЁ(1)жївЊЦРМлжИБъ
ЁЁЁЁЂйжївЊЦРМлжИБъЃКЪѕКѓ12ИідТЦРЗжЗжЪ§ЁЂЪѕЧАЪѕКѓ12ИідТЦРЗжИФБфЗжЪ§ЁЂЛђЪѕКѓ12ИідТЦРЗж“гХСМТЪ”(МДЃКжУЛЛїХЙиНкМйЬхКѓЦРЗжДяЕНгХЁЂСМЕФБШР§ЁЃ)
ЁЁЁЁЂкЦРЗжЗНЗЈЃКГѕДЮжУЛЛаЭЁЂЗаоаЭїХЙиНкПЩВЩгУHarrisЦРЗж(МћИН3)ЃЌHarrisЦРЗжТњЗж100ЗжЃЌ90ЗжвдЩЯЮЊгХЃЌ80—89ЗжЮЊСМЃЌ70—79ЗжЮЊПЩЃЌаЁгк70ЗжЮЊВю;жзСіаЭїХЙиНкПЩВЩгУMSTSЦРЗж(МћИН4)ЃЌИУЯЕЭГТњЗж30ЗжЃЌ≥24ЗжЖЈвхЮЊгХЃЌ18—23ЗжЖЈвхЮЊСМЃЌ12—17ЗжЖЈвхЮЊжаЃЌ≤11ЗжЖЈвхЮЊВюЁЃ
ЁЁЁЁ(2)ДЮвЊЦРМлжИБъ
ЁЁЁЁЂйXЩфЯпЦНЦЌВЮЪ§ЃКЩъЧыШЫгІжЦЖЈгАЯёбЇГЩЙІЕФБъзМВЂНјааЦРМлЁЃЭЌЪБЙизЂМйЬхжмЮЇЭИССЯпЕФаЮГЩ(ЙЧЫЎФржЪСПЁЂвьЮЛЙЧЛЏЁЂМйЬхЫЩЖЏ)ЁЂЙЧжЪШмНтЁЂМйЬхЮЛжУБфЛЏ(ЭЗОБМйЬхЯТГСЁЂїХОЪМйЬхЕФЧуаБНЧЖШБфЛЏЁЂїХОЪМйЬхЕФФквЦЛђЩЯвЦЁЂЙЩЙЧБњОБИЩНЧЕФБфЛЏгаЮоФкЗЛђЭтЗЕШ)ЁЂЙиНкЭбЮЛЕШЕФЗЂЩњТЪЁЃ
ЁЁЁЁЂкЩњДцТЪЃКИљОнШЁГіЛђепАќРЈШЁГіїХЙиНкМйЬхЕФШЮКЮвЛВПЗжЕФВЁР§РДМЦЫуМйЬхЕФЩњДцТЪЁЃЗЂЩњ1Р§гыЩЯЪіВњЦЗжЪСПЯрЙиЕФбЯжиВЛСМЪТМўЃЌХаЖЈСйДВЪдбщЪЇАмЁЃ
ЁЁЁЁЂлВЛСМЪТМўЗЂЩњТЪЁЃ
ЁЁЁЁЂмВЂЗЂжЂЗЂЩњТЪЃКаыЖдВЂЗЂжЂЗЂЩњЕФРраЭЁЂЪ§СПЁЂБШР§НјааЭГМЦЗжЮіЃЌВЂТлжЄЦфгыжВШыїХЙиНкМйЬхЕФЯрЙиадЁЃ
ЁЁЁЁ7.ЖдееВњЦЗЕФбЁдё
ЁЁЁЁЖдееВњЦЗгІбЁдёФПЧАСйДВе§ЙуЗКЪЙгУЕФЁЂЖдЯргІЪЪгІжЄЕФСЦаЇвбБЛжЄЪЕВЂЕУЕНЙЋШЯЕФРДдДгкЕЅвЛГЇМвЩњВњЕФЭЌвЛЯЕЭГВњЦЗЁЃЖдееВњЦЗЕФВФСЯЁЂЩшМЦЁЂЪЪгІжЄгыЪдбщВњЦЗОпгаПЩБШадЁЃЩъЧыШЫгІЬсЙЉЖдееВњЦЗЕФбЁдёвРОнЁЃ
ЁЁЁЁ8.бљБОСПЕФЙРЫу
ЁЁЁЁвНСЦЦїаЕВњЦЗзЂВсЩъЧыШЫгІЬсЙЉбљБОСПзувдЦРМлЫљЩъБЈВњЦЗАВШЋадКЭгааЇадЕФЭГМЦбЇжЄОнЃЌАќРЈвдЯТФкШнЃКЖдеезщгыЪдбщзщжївЊЦРМлжИБъЯрЭЌЪдбщЬѕМў(ЭЌбљЕФЪЪгІжЄШЫШКЁЂжЮСЦЪБМфЁЂЫцЗУЪБМфЕШ)ЯТЕФдЄЦкСЦаЇЁЂдЄЦкЕФзщМфВювьЁЂЯджјадЫЎЦН(α)ЁЂАбЮеЖШ(β)ЁЂдЄЦкЪЇЗУТЪЁЂЫљгУЕНЕФбљБОСПМЦЫуЙЋЪНЁЂЫљгаЕФМЦЫуЙ§ГЬМАЪЙгУЕФЭГМЦбЇШэМўЁЂв§гУЕФВЮПМЮФЯзЕШЁЃ
ЁЁЁЁЩъЧыШЫгІИљОнВњЦЗЕФадФмжИБъбЁдёЖдееЦЗЃЌВЂВЩгУОЕфЕФЭГМЦбЇЗНЗЈМАЙњФкЭтЙЋШЯЕФЭГМЦбЇШэМўМЦЫубљБОСПЁЃ
ЁЁЁЁР§ШчЃК
ЁЁЁЁМйЩшФГЫцЛњЖдееЗЧСгаЇСйДВЪдбщЃЌИљОнЮФЯзБЈЕРЃКЭЌРрВњЦЗЕФгХСМТЪЮЊ95%ЁЂСйДВШЯПЩЕФЗЧСгаЇНчжЕЮЊ10%ЃЌдђдкЫЋВрЯджјадЫЎЦН0.05ЁЂАбЮеЖШ80%ЁЂЭбТфТЪ10%ЪБЃЌУПзщашвЊ84Р§ЁЃ
ЁЁЁЁИУбаОПЮЊЫцЛњЖдееЗЧСгаЇСйДВЪдбщЃЌжївЊЦРМлжИБъЪЧЪѕКѓ12ИідТHarrisЦРЗжЁЃИљОнЮФЯзБЈЕРЃЌЖдееВњЦЗЕФЦРЗжЮЊ90±10ЗжЃЌСйДВШЯПЩЕФЗЧСгаЇНчжЕЮЊ5ЃЌдђдкЫЋВрЯджјадЫЎЦН0.05ЁЂАбЮеЖШ80%ЁЂЭбТфТЪ10%ЪБЃЌУПзщаш70Р§ЁЃ
ЁЁЁЁИУбаОПЮЊЕЅзщФПБъжЕЪдбщЃЌжївЊЦРМлжИБъЪЧЪѕКѓ12ИідТHarrisЦРЗж“гХСМТЪ”ЁЃИљОнЮФЯзБЈЕРЃЌбаОПВњЦЗЕФгХСМТЪЮЊ95%ЃЌСйДВШЯПЩЕФФПБъжЕЮЊ85%ЃЌдђдкЫЋВрЯджјадЫЎЦН0.05ЁЂАбЮеЖШ80%ЁЂЭбТфТЪ10%ЪБЃЌаш87Р§ЁЃ
ЁЁЁЁОіЖЈбљБОСПЕФЙиМќвђЫигаЃКбаОПРраЭЁЂжївЊЦРМлжИБъЁЂЖдеезщгыЪдбщзщжївЊЦРМлжИБъЕФдЄЦкСЦаЇЁЂЗЧСгаЇНчжЕЛђФПБъжЕЁЂЯджјадЫЎЦН(α)ЁЂАбЮеЖШ(β)ЁЂдЄЦкЪЇЗУТЪЕШЁЃ
ЁЁЁЁШєНјааЫцЛњЖдееЗЧСгаЇЪдбщЃЌдђашУїШЗЖдееВњЦЗдЄЦкСЦаЇКЭСйДВШЯПЩЕФЗЧСгаЇНчжЕ;ШєНјааЕЅзщФПБъжЕЪдбщЃЌдђашУїШЗЪдбщВњЦЗдЄЦкСЦаЇКЭСйДВШЯПЩЕФФПБъжЕЁЃ
ЁЁЁЁ9.ШЫПкЭГМЦбЇКЭЛљЯпЬиеї
ЁЁЁЁ(1)ШЫПкЭГМЦбЇзЪСЯЃКШчадБ№ЁЂФъСфЁЂУёзхЁЂЩэИпЁЂЬхжиЕШ;
ЁЁЁЁ(2)СйДВСЦаЇЯрЙиЕФЛљЯпЪ§ОнЃКПМТЧвђЫиАќРЈМВВЁЕФеяЖЯЁЂЗжЦкЁЂЗжМЖМАгАЯёбЇВЮЪ§ЁЂАщЫцМВВЁЁЂЦфЫћЙиНкЮЪЬтЁЂЗчЯевђЫиЁЂЗХЩфбЇУшЪі(CCDЁЂЭШГЄЁЂЦЋаФОрЕШ);
ЁЁЁЁ(3)КЯВЂжЂЃКЪЧЗёгаЙЧжЪЪшЫЩЁЂгЊбјВЛСМ(ИЦЁЂСзЁЂЕААзжЪЁЂЬњ)ЁЂЦЖбЊЁЂМЄЫиШБЗІ(ЩњГЄМЄЫиЁЂМззДХдЯйЫиЕШ)ЁЂЗХЩфжЮСЦЁЂЪжЪѕЪЗЁЂЬЧФђВЁЪЗЁЂИпбЊбЙЁЂЙкаФВЁЁЂЗЮЙІФмЧщПіЁЂУтвпбЇМВВЁЕШЁЃ
ЁЁЁЁ10.ЭГМЦЗжЮіЗНЗЈ
ЁЁЁЁгІУїЪООпЬхЕФЭГМЦЗжЮіЗНЗЈвдМАЭГМЦЗжЮіШэМўМААцБОЁЃ
ЁЁЁЁЪ§ОнЗжЮіЪБгІПМТЧЪ§ОнЕФЭъећадЃЌЫљгаЧЉЪ№жЊЧщЭЌвтВЂЪЙгУСЫЪмЪдВњЦЗЕФЪмЪдепБиаыФЩШыЗжЮіЁЃЪ§ОнЕФЬоГ§ЛђЦЋвЦЪ§ОнЕФДІРэБиаыгаПЦбЇвРОнКЭЯъЯИЫЕУїЁЃ
ЁЁЁЁвНСЦЦїаЕСйДВЪдбщЕФЪ§ОнЗжЮігІЛљгкВЛЭЌЕФЗжЮіМЏЃЌЭЈГЃАќРЈШЋЗжЮіМЏ(Full Analysis SetЃЌFAS)ЁЂЗћКЯЗНАИМЏ(Per Protocol SetЃЌPPS)КЭАВШЋМЏ(Safety SetЃЌSS)ЃЌбаОПЗНАИжагІУїШЗИїЗжЮіМЏЕФЖЈвхЁЃШЋЗжЮіМЏжаЭбТфВЁР§ЃЌЦфжївЊбаОПжеЕуЕФШБЪЇжЕЕФЬюВЙЗНЗЈЕШгІдкЗНАИжаЪТЯШгшвдЫЕУїЃЌВЂНјааВЛЭЌЗжЮіВпТдЕФСщУєЖШЗжЮіЃЌвдЦРМлШБЪЇЪ§ОнЖдбаОПНсЙћЮШЖЈадЕФгАЯьЁЃ
ЁЁЁЁжївЊбаОПжеЕужИБъЕФЗжЮігІЭЌЪБдкШЋЗжЮіМЏКЭЗћКЯЗНАИМЏЩЯНјаа;АВШЋаджИБъЕФЗжЮігІЛљгкАВШЋМЏЁЃ
ЁЁЁЁСйДВЪдбщЪ§ОнЕФЗжЮігІВЩгУЙњФкЭтЙЋШЯЕФОЕфЭГМЦЗжЮіЗНЗЈЁЃСйДВЪдбщЗНАИгІИУУїШЗЭГМЦМьбщЕФРраЭЁЂМьбщМйЩшЁЂХаЖЈСЦаЇгаСйДВвтвхЕФНчжЕ(ЗЧСгаЇНчжЕ)ЕШЃЌНчжЕЕФШЗЖЈгІгавРОнЁЃ
ЁЁЁЁЖдгкжївЊбаОПжеЕуЃЌЭГМЦНсЙћашВЩгУЕуЙРМЦМАЯргІЕФ95%ПЩаХЧјМфНјааЦРМлЁЃВЛФмНіНЋpжЕзїЮЊЖджївЊбаОПжеЕуНјааЦРМлЕФвРОнЁЃ
ЁЁЁЁЖдЪдбщЦкМфЗЂЩњЕФЫљгаВЛСМЪТМўЕФжжРрЁЂбЯжиГЬЖШЁЂЗЂЩњЦЕТЪМАгыЪдбщВњЦЗЕФЙиЯЕНЋСаБэУшЪіЁЃ
ЁЁЁЁЩъЧыШЫгІЬсЙЉЛљгкЫљгазЂВсСйДВЪдбщЪ§ОнЕФЭГМЦЗжЮіБЈИцЃЌвдБуСйДВЪдбщзщГЄЕЅЮЛИљОнДЫБЈИцзЋаДСйДВЪдбщзмНсБЈИцЁЃ
ЁЁЁЁ11.ЦфЫћ
ЁЁЁЁвбШЁЕУвНСЦЦїаЕВњЦЗзЂВсжЄЃЌЧвЪЪгУЗЖЮЇЮЊГѕДЮжУЛЛЕФВњЦЗЃЌдкаТдіВњЦЗгыИУзЂВсжЄВњЦЗЪєгкЭЌвЛзЂВсЕЅдЊЕФЧщПіЯТЃЌдіМгЪЪгУгкЗаоЕФВњЦЗаЭКХЙцИёМАЪЪгУЗЖЮЇЃЌПЩдкЭЌЪБТњзуШчЯТЬѕМўЕФЧщПіЯТгшвддіМгЃКШЁЕУзЂВсжЄТњ2ФъЁЂШЁЕУзЂВсжЄКѓЯњЪлЕФВњЦЗЫцЗУЪ§СПжСЩйДяЕН100Р§ЧвВњЦЗжВШы12ИідТКѓгХСМТЪВЛЕЭгк95%ЁЂЭъГЩЯрЙиЙІФмадЪдбщЁЂБИЦыЯрЙизЂВсЮФМўЁЃ
ЩюлкКшдЖвНСЦЦїаЕзЩбЏЗўЮёЙЋЫО ЪЧвЛМвММЪѕзЈвЕЕФвНСЦЦїаЕзЩбЏЗўЮёЙЋЫОЃЌзЈзЂЬсЙЉШЋЙњИїЕиШчЃКЩюлкЁЂЙужнЁЂЖЋнИЁЂжаЩНЁЂЗ№ЩНЁЂГБжнЁЂЫГЕТЁЂЩЯКЃЁЂЮїАВЁЂжиЧьЁЂГЩЖМЁЂАВЛеЁЂНЫеЁЂеуНЕШжЊУћГЧЪаЕФвНСЦЦїаЕСьгђММЪѕзЩбЏЗўЮёЁЃЩюлкКшдЖвНСЦЦїаЕзЩбЏзЈвЕЗўЮёгкЃКвНСЦЦїаЕВњЦЗзЂВсжЄДњАьРэзЩбЏЁЂДњАьвНСЦЦїаЕЩњВњаэПЩжЄЁЂвЛРрвНСЦЦїаЕВњЦЗБИАИДњАьЁЂвНСЦЦїаЕОгЊаэПЩжЄДњАьЁЂЖўРрвНСЦЦїаЕОгЊБИАИЁЂНјПквНСЦЦїаЕзЂВсЁЂCEШЯжЄЁЂISO13485ШЯжЄЁЂFDAзЂВсЁЂFDAШЯжЄЁЂСйДВЪдбщЁЂвНСЦЦїаЕжЪСПЙмРэЬхЯЕШЯжЄМАЬхЯЕНЈСЂгыЙ§ГЬШЗШЯЮФМўНЈСЂ(ISO9001, ISO13485, GMP, CEЃЌQSR820ЃЌCMDCAS);зЂВсЁЂГіПкжЄЁЂздгЩЯњЪлжЄАьРэЁЂВњЦЗММЪѕвЊЧѓжЦЖЉЁЂММЪѕЮФМўЁЂСйДВЪдбщМАУтСйДВЭЌРрВњЦЗЖдБШзЪСЯБраДЁЂзЂВсзЪСЯБраДИЈЕМЁЂЕчДХМцШнећИФЁЂвНСЦЦїаЕЙуИцХњЮФЩъБЈЕШЬсЙЉвЛеОЪНЗўЮёЙЋЫОЃЌЛЖгФњзЩбЏгыКЯзїЃЁ
вНСЦЦїаЕзЂВсзЩбЏЗўЮёЙЋЫО
вНСЦЦїаЕзЪбЖ
- 2023ФъвНСЦЦїаЕзЂВсСйДВЦРМлзЪСЯдѕУДБраД?
- МЄЙтжЮСЦЩшБИЭЌЦЗжжСйДВЦРМлзЂВсЩъБЈзЪСЯгаФФ
- 2023ФъаТаоЖЉЁЖУтгкСйДВЦРМлвНСЦЦїаЕФПТМЁЗ
- ПкЧЛжжжВЪжЪѕЕМКНЖЈЮЛЯЕЭГЭЌЦЗжжСйДВЦРМлзЂВс
- вНСЦЦїаЕСйДВЪдбщЪ§ОнЕнНЛвЊЧѓзЂВсЩѓВщжИЕМд
- 2021ФъСаШыУтгкСйДВЦРМлвНСЦЦїаЕФПТМВњЦЗЖдБШ
- 2021ФъЁЖУтгкСйДВЦРМлвНСЦЦїаЕФПТМЁЗ
- вНСЦЦїаЕбЊЙмФкЕМЙмВњЦЗзЂВсЭЌЦЗжжЖдБШСйДВЦР
- ЁЖгІгУФЩУзВФСЯЕФвНСЦЦїаЕАВШЋадКЭгааЇадЦРМл
- вНСЦЦїаЕїХЙиНкМйЬхВњЦЗзЂВсШчКЮбЁдёСйДВЦРМл
 дкЯпПЭЗў
дкЯпПЭЗў